

Ciao a tutti! Sono Vincenzo Lomonaco, ricercatore all’Università di Pisa. Nella puntata di oggi discuteremo di AlphaFold 3, il recentissimo modello di intelligenza artificiale rilasciato da Google DeepMind e Isomorphic Labs, che ha l’obiettivo di prevedere con precisione la struttura di proteine, DNA, RNA, ligandi e altro ancora, nonché di comprendere come interagiscono tra loro.
Infatti, ci auguriamo che questa scoperta possa migliorare significativamente la nostra comprensione del mondo biologico e la scoperta di nuovi farmaci.
Ma facciamo un passo indietro.
Che cos’è il problema del ripiegamento proteico o protein folding? Le proteine, come sappiamo, svolgono funzioni essenziali nel nostro organismo.
La loro attività biologica è strettamente legata alla struttura tridimensionale che deriva dal complesso processo di ripiegamento proteico. Il termine proteina si riferisce a composti contenenti azoto costituiti da una combinazione di amminoacidi legati da un legame peptidico. Queste molecole sono responsabili del supporto strutturale cellulare, della protezione immunitaria, della catalisi enzimatica, della trasduzione del segnale cellulare per la regolazione della trascrizione e della traduzione.
Quindi la funzione biologica di una proteina dipende dalla sua struttura terziaria e quaternaria, che deriva dal ripiegamento di una sequenza polipeptidica.
In sintesi, la struttura terziaria definisce la forma di una singola catena di amminoacidi, mentre la struttura quaternaria riguarda l’assemblaggio di più catene piegate per formare una proteina funzionale. Ecco, comprendere il ripiegamento proteico rappresenta uno degli obiettivi più importanti della biologia strutturale.
Sperimentalmente le strutture proteiche terziarie vengono scoperte mediante tecniche come la cristallografia a raggi X, la risonanza magnetica nucleare e la criomicroscopia elettronica.
Tuttavia, queste tecniche fondamentali per determinare la struttura delle proteine presentano significative limitazioni.
La cristallografia, per esempio, richiede cristalli di buona qualità e può avere difficoltà con proteine di grandi dimensioni o non cristalline.
La risonanza magnetica nucleare, invece, è efficace principalmente per proteine di piccole e medie dimensioni e richiede spesso marcature isotopiche oltre a essere difficile da interpretare per proteine più grandi.
Infine, la criomicroscopia ha limitazioni della risoluzione e richiede elaborazioni computazionali piuttosto complesse.
In alcuni casi, è necessario combinare queste tecniche per ottenere una comprensione completa della struttura proteica.
Prima dell’avvento di AlphaFold, i ricercatori hanno sviluppato metodi per prevedere la struttura proteica combinando approcci basati su modelli di omologia e modellazione libera.
La modellazione di omologia si basa sul fatto che le proteine con una sequenza simile tendono ad avere una struttura simile.
Utilizzando questo principio, si costruiscono modelli di proteine sconosciute basandosi su proteine conosciute anche a livello di struttura tridimensionale, che vengono chiamate templates e che hanno una sequenza ovviamente simile.
D’altra parte, la modellazione libera non utilizza un template, ma si basa su principi fisici e chimici fondamentali per predire la struttura tridimensionale di una proteina partendo dalla sua sequenza amminoacidica.
Entrambi gli approcci hanno i loro vantaggi e limitazioni e sono utilizzati spesso in combinazione.
Per questo motivo, per ottenere predizioni accurate delle strutture proteiche, nel 2020 AlphaFold 2, basato sulla prima versione prototipale del 2016, se non erro, ha realizzato una svolta fondamentale nella predizione della struttura proteica.
Finalmente, a partire da una semplice sequenza amminoacidica, è stato possibile ottenere la struttura tridimensionale corrispondente della proteina in modo estremamente efficiente tramite tecniche di apprendimento automatico.
Ad oggi, già milioni di ricercatori in tutto il mondo hanno utilizzato AlphaFold 2 per fare scoperte diverse in aree che includono i vaccini antimalarici, i trattamenti contro il cancro e la progettazione di enzimi.
Tant’è che a sua volta è stato citato il paper, l’articolo di riferimento, più di 20.000 volte e il suo impatto scientifico è stato riconosciuto attraverso diversi premi. Il più recente, il Breakthrough Prize in Life Sciences.
AlphaFold 3 rappresenta un notevole avanzamento rispetto alla sua versione precedente AlphaFold 2.
Nel campo della predizione della struttura proteica, grazie a nuove tecniche e algoritmi, offre infatti una maggiore precisione e in alcuni casi maggiore anche del 50% rispetto al modello precedente e affidabilità nelle predizioni della struttura tridimensionale delle proteine, superando le limitazioni della versione precedente.
I principali vantaggi di AlphaFold 3 includono una migliore capacità di predire la struttura di proteine di dimensioni più grandi e complesse, una maggiore accuratezza nei dettagli locali della struttura e una maggiore robustezza nel predire le interazioni proteina proteina.
Gli avanzamenti tecnologici che hanno reso questo possibile includono l’utilizzo di, ovviamente, una più ampia gamma di dati sperimentali, l’ottimizzazione di algoritmi di apprendimento automatico e l’implementazione di tecniche innovative per migliorare la qualità delle predizioni a valle in una fase successiva di rifinitura.
A titolo di esempio, in questa immagine vediamo la predizione circa la struttura tridimensionale di una struttura complessa denominata 7R6R, composta da una proteina in blu ed una doppia elica di DNA in rosa.
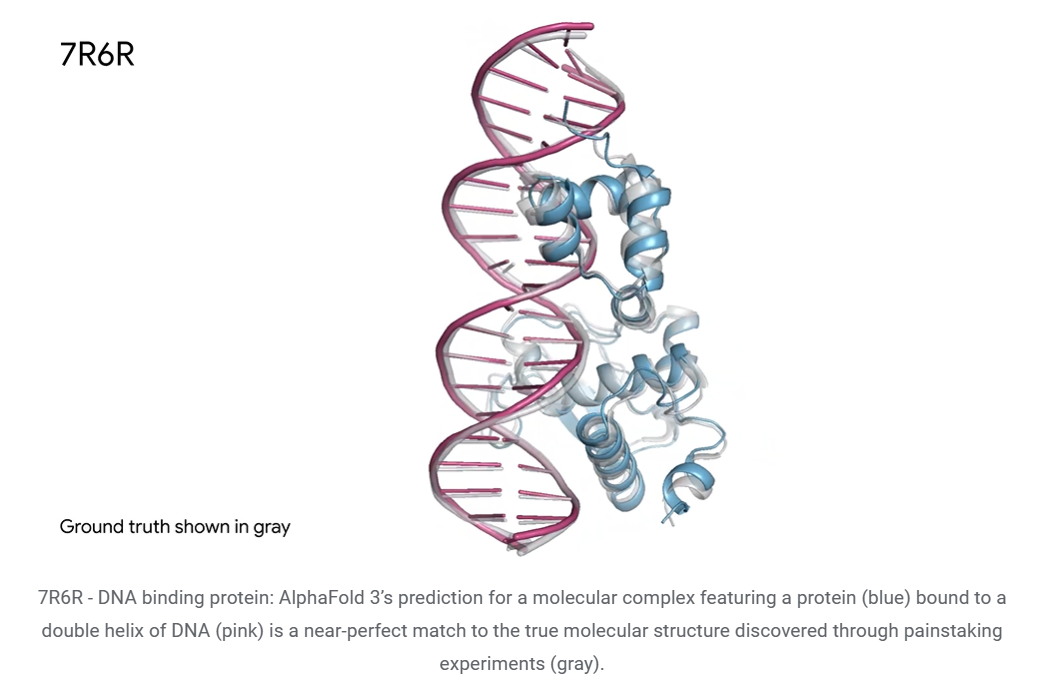
Se ci soffermiamo un attimo di più sull’immagine, notiamo anche una sorta di ombra in grigio chiaro.
Ecco, questa rappresenta la reale struttura verificata in laboratorio e che ci consente di valutare la bontà della nostra predizione.
È chiaro che in questo caso la rappresentazione colorata è quella predetta dalla foto solo a partire dalla sequenza amminoacidica risulta sostanzialmente sovrapposta a quella veritiera in grigio, evidenziando un’ottima capacità predittiva del modello.
Contestualmente al lancio del nuovo modello di AI, attestato da un recente articolo pubblicato su Nature, che vi invito a consultare se volete saperne di più, il gruppo di ricercatori capeggiato da Demis Hassabis ha anche lanciato in fase beta sperimentale un AlphaFold server con l’idea di offrire uno strumento molto facile da usare, basato su un servizio web del browser per offrire a tutti la possibilità di utilizzare questo modello di eye senza scrivere una linea di codice.
In questa immagine vediamo uno screenshot dell’AlphaFold Server che ho testato proprio questa mattina, provando a inserire la sequenza amminoacidica dell’insulina e questo è il risultato.
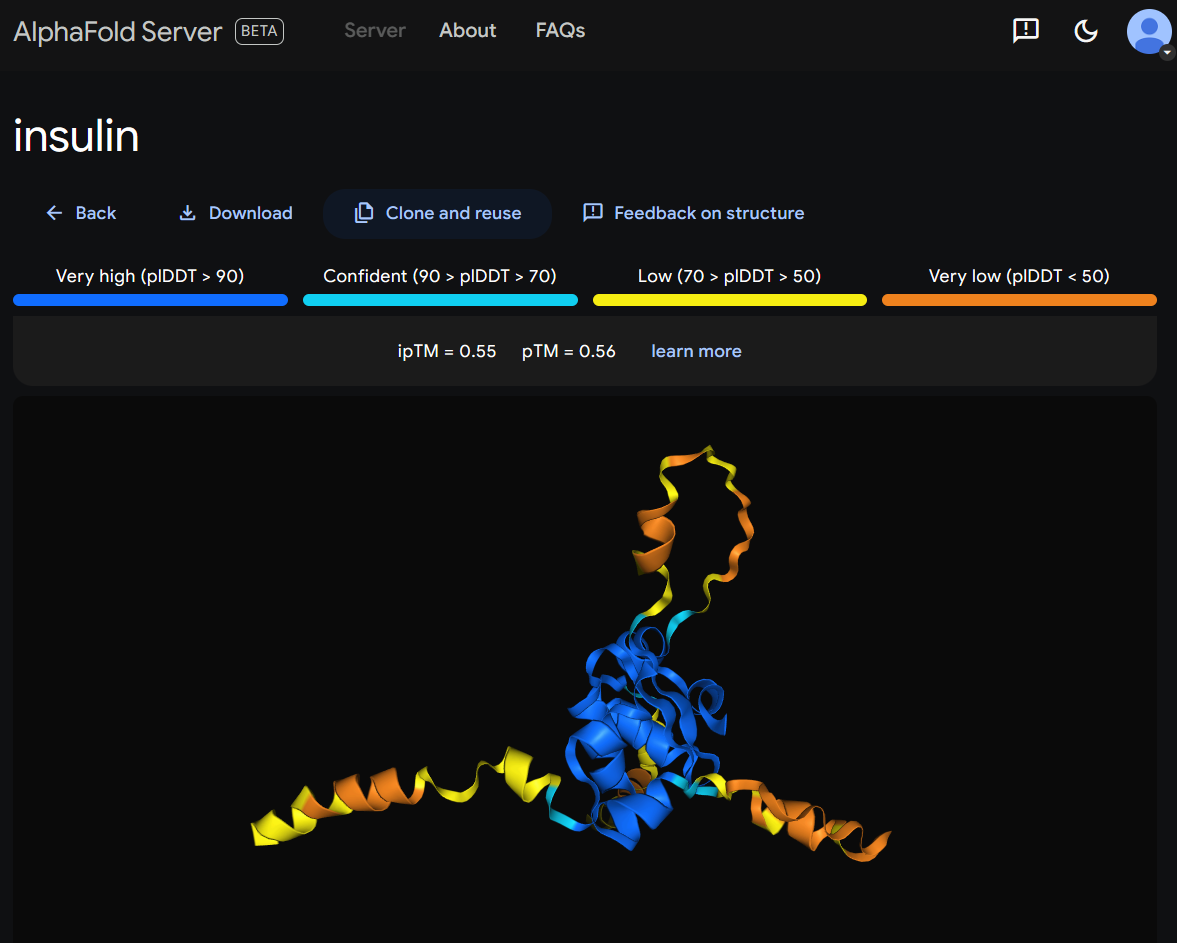
Come vediamo, Alpha Fold in pochi secondi ha generato per me la struttura tridimensionale della sequenza e mi ha anche fornito tutta una serie di dati scaricabili sulla confidenza che ha nelle sue predizioni.
Per esempio è molto più confidente della parte centrale della proteina in blu, mentre è meno confidente in giallo e arancione riguardo le porzioni più esterne.
Per riassumere, AlphaFold sta rivoluzionando sicuramente il campo della biologia molecolare e rappresenta uno dei successi più concreti dell’intelligenza artificiale moderna.
Nel futuro vedremo sempre più applicazioni di AlphaFold e modelli analoghi per la scoperta di nuovi farmaci, l’ingegneria proteica, la diagnosi medica, soprattutto nell’ottica di questa nuova era della medicina di precisione.
Ciao! Alla prossima puntata di Le Voci dell’AI






{kind=link}